DISpro 1.0 is a software which predict the disordered regions from protein sequences.
-----------------------------------------------------------------------------------------
DoBo is a sequence based protein domain boundary predictor. It leverages evolutionary information contained in multiple sequence alignments to identify potential domain boundary sites. These candidate sites are then classified using a support vector machine. Predicted domain boundary sites are finally scored and a confidence value provided.
-----------------------------------------------------------------------------------------
What is DoBo?
DoBo (Domain Boundary) is a tool to identify domain boundaries from sequence. It works by combining the classification power of machine learning with domain boundary signals embedded in multiple sequence alignments. More specifically, a multiple sequence alignment is generated for a query sequence and signals are generated. A domain boundary signal is defined as a gap which starts at either end of a protein sequence from the MSA and continues for at least 45 residues. Once signals are identified, they can be classified using a Support Vector Machine. Fig. 1. depicts signal detection.
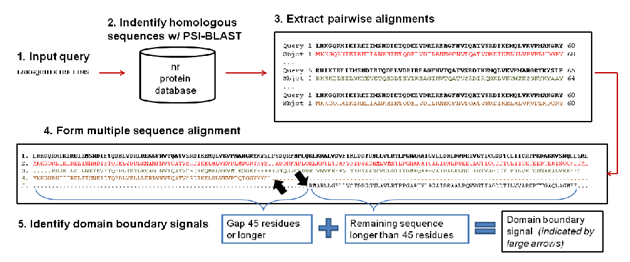
Fig. 1.
-----------------------------------------------------------------------------------------
Why should I use DoBo?
DoBo is an ab-initio protein domain boundary prediction package. As it does not need template or domain information from homologous proteins, it lends itself well to predicting domain boundaries in novel sequences. It also allows boundaries to be predicted a varying confidence values. Setting the confidence level to 80% means that on average, 80 percent of the domain boundary predictions will be near (+/- 20 residues) a true domain boundary.
-----------------------------------------------------------------------------------------
Why was I told to lower the decision threshold? Why weren't boundaries predicted?
DoBo allows a user to set a confidence threshold on predicted domain boundaries. If a signal is generated at a residue location but is not scored about the confidence threshold, a domain boundary will not be predicted at that site. To view all signals generated, you can view the "signals.lst" file. A sample is shown below. It contains three columns corresponding to the residue location, signal score and confidence value of the site.
712 -0.613048 0.59
705 -0.616138 0.59
459 -0.627498 0.59
493 -0.651736 0.58
1207 -0.677879 0.57
1298 -0.711237 0.56
1059 -0.714784 0.56
1032 -0.710711 0.56
Other reasons why predictions might not be made are sequence length and lack of similar sequences. Due to the implentation, the minimum sequence length is 90 residues. Any sequence less than that length will not generate signals.
In very rare cases, it may be difficult to generate a multiple sequence alignment for a query protein. To see if this is the case, you may inspect the MSA generated for your query by following the link at the bottom of the results page.
-----------------------------------------------------------------------------------------
What should I cite?
Please cite: J. Eickholt, X. Deng, and J. Cheng. DoBo: Protein Domain Boundary Prediction by Integrating Evolutionary Signals and Machine Learning. BMC Bioinformatics. 12:43, 2011.
For any other questions or concerns, contact Jesse Eickholt at jlec95@ mail.mizzou.edu.
|