Thanks for using APOLLO web server. The following is the quality assessment report for your recent submission to our web server.
If you have submitted a single model, APOLLO evaluates and outputs its absolute quality. If you have submitted multiple models, APOLLO outputs (1) the absolute quality score, (2) pairwise GDT-TS score, (3) refined absolute scores, and (4) refined pairwise Q score, respectively. If more than five models have absolute scores reported, local quality scores are attached with this email, which are generated by refining absolute scores. Please be aware that the quality scores generated using pairwise comparisons (2, 4) only make sense when the number of models reaches a certain number, and we recommend this number to be at least 20.
Detailed explanations about the methods and outputs can be found at here.
APOLLO model evaluation report: (machine readable local-quality)(Machine-readable predicted secondary structure, solvent accessibilities, and contact map)
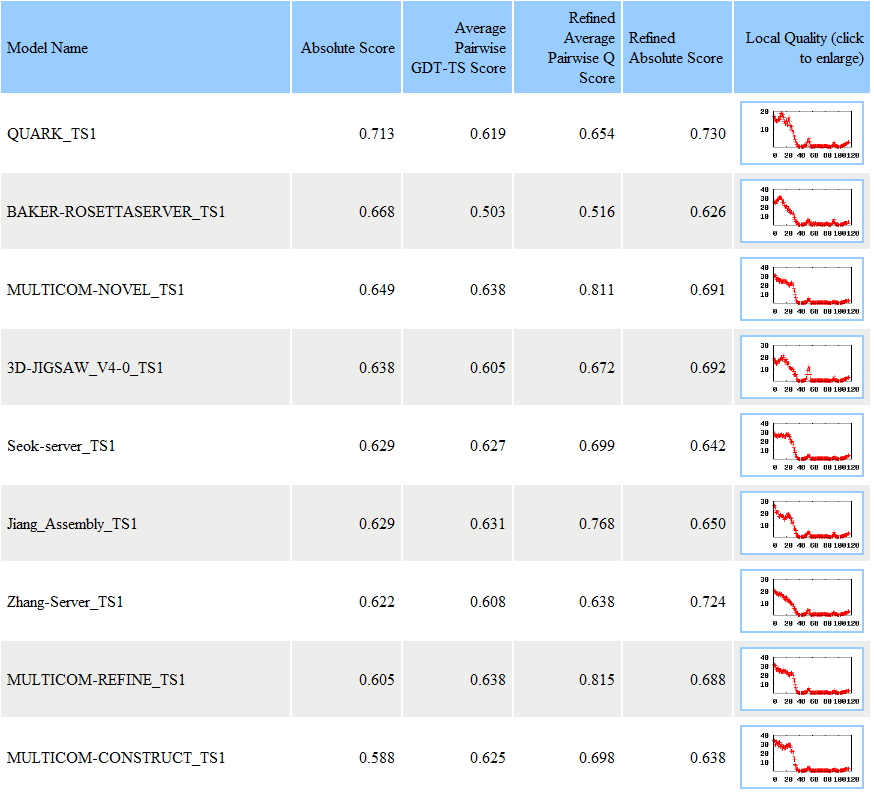
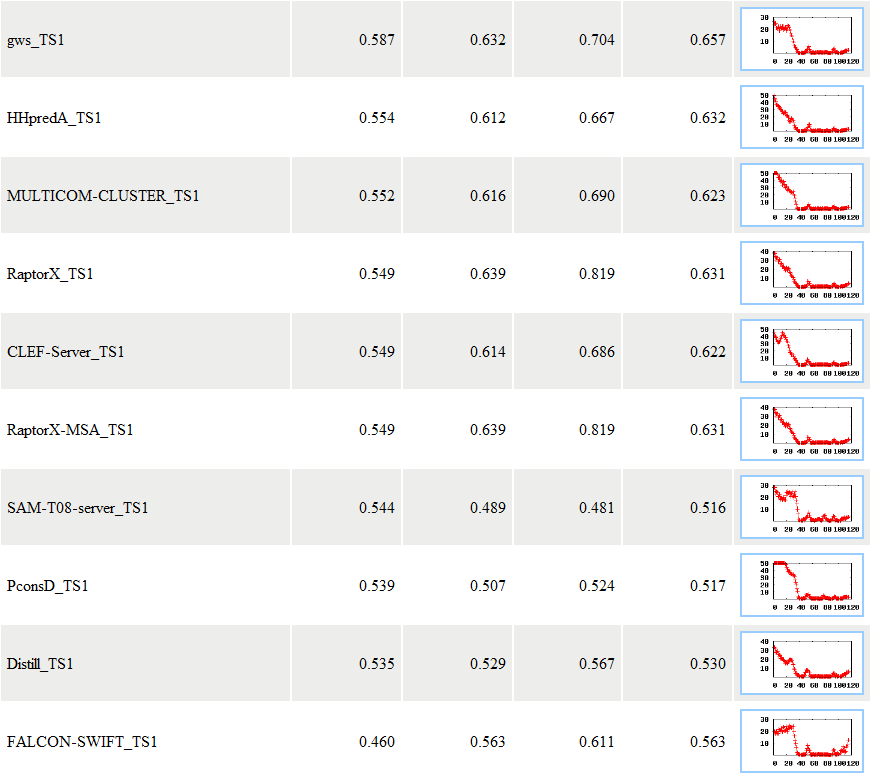
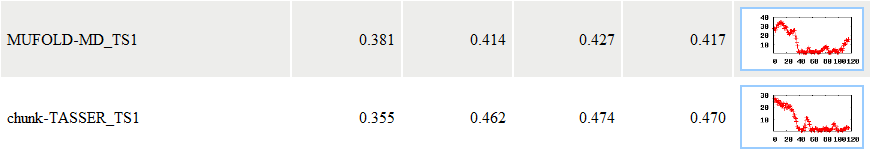
In this example, APOLLO reported the evaluation results for seven models. For each model, APOLLO sent three scores: the absolutely quality score - the predicted GDT-TS score between a model and the unknown true tertiary structure, the average pairwise GDT-TS score, and the average pairwise Q score. A GDT-TS score within the range [0, 1] is a standard measure for quantifying structural similarity. An absolute GDT-TS score of greater than 0.4 often indicates that the model has the same topology as the true structure. For example, for model MULTICOM-NOVEL_TS1, its absolute quality score is 0.649, the average pairwise GDT-TS score is 0.638, and the average pairwise Q score is 0.811. The models are ranked based on the absolute quality score. The absolute quality score is calculated based on a single model using Support Vector Machine. The average correlation between the GDT-TS scores predicted by this method and the models' true GDT-TS scores can reach 0.67 when blindly tested on CASP9 targets. Details of the method and its evaluation can be found at here (as MULTICOM-NOVEL) or at our publication:
Wang, Z., Tegge, A.N., and Cheng, J. (2009) Evaluating the Absolute Quality of a Single Protein Model using Structural Features and Support Vector Machines. Proteins, 75, 638-647. (pdf)
The average pairwise GDT-TS score is calculated based on a pool of models of the same target protein. Given a pool of models, it performs a structural alignment between each pair of models, and generates a GDT-TS score between two models. For a model, it calculate the average GDT-TS score of all the comparisons between this model and the others. This average pairwise GDT-TS score is used as the predicted quality score. Pairwise comparision based mothod works best when the number of models reachs a big number and with various qualities. For example, the average correlation between the predictions generated from this pairwise GDT-TS based evaluation method and the models' true GDT-TS scores can reach 0.92 when it was blindly applied on CASP9's more than 100 targets, each of which contains ~300 models generated by 140 protein tertiary structure prediction groups from all over the world. Detailed descriptions can be found here (as MULTICOM-CLUSTER)
The refined average pairwise Q score is calculated based on a pool of models of the same target protein. Instead of using the GDT-TS score between each pair of two models, it calculates the Q score between each pair of two models, and then calculates its average. The models are ranked by this pairwise Q scores and top five models are selected as reference models. For each of the model, it uses the tool TM_align (http://zhang.bioinformatics.ku.edu/TM-align/) to superimpose it with each one of the reference models, which outputs a distance score between each pair of amino acids. The average distance score over the 5 reference models are used as the local quality of an amino acid position. Therefore, if the pool has less than 5 models, local quality scores will not be provided. The detailed descriptions about how Q score is calculated can be found at here (as MULTICOM-CONSTRUCT) or:
McGuffin, L. & Roche, D. (2010). Rapid model quality assessment for protein structure predictions using the comparison of multiple models without structural alignments. Bioinformatics 26, 182-188.
The Refined Absolute Score is generated by the same refinement precedure mentioned above. The only difference is that here it is a refinement on the absolute scores. All of the above predicted quality scores range between [0, 1], whereas 1 indicates a perfect model with the same structure as native, and 0 indicating no similarity with the native structure. The absolute quality only uses a single model as input, however, the other two quality scores generated based on pairwise comparisons only make sense when the model pool reach a certain size. For a pool which contains less than 20 models, or a pool that only contains bad quality models, we do not think these two scores make much sense.
APOLLO also generates a machine-readable file, which contains the local quality assessment of a pool of models, i.e., it gives a predicted distance between the model and the native structure on each of the amino acid position of a model. It ranks all the models in the pool based on the absolute qualities, and then selects top 5 models as reference models. For each of the model, it uses the tool TM_align (http://zhang.bioinformatics.ku.edu/TM-align/) to superimpose it with each one of the reference models, which outputs a distance score between each pair of amino acids. The average distance score over the 5 reference models are used as the local quality of an amino acid position. Therefore, if the pool has less than 5 models, local quality scores will not be provided. The local quality assessment report follows the format of CASP (Critical Assessment of Techniques for Protein Structure Prediction), which can be found at here. For every model, it ouputs a string of numbers, whereas the first number is the global quality score and the following are the local scores. For example, in the example shown below, the global score for model "MidwayFoldingServer_TS3" is 0.41846, and following it are the local quality scores for each amino acid residue. The method and evaluations can be found here (as MULTICOM-REFINE)
MidwayFoldingServer_TS3 0.41846 16.11 18.4 16.53 16.71 18.32 18.43 17.06 16.72 16.25 17.23 16.54 14.99 16.77 16.78 15.65 13.66
14.84 14.91 13.83 12.41 11.61 11.06 11.93 11.81 12.38 12.84 12.36 13.43 12.68 12.26 12.16
13.34 12.61 12.38 13.23 13.08 12.4 9.93 7.72 6.3 4.8 3.66 3.0 1.68 1.88 2.51
3.61 5.8 7.51 9.64 9.45 9.58 8.06 7.97 7.65 7.62 8.18 8.97 8.66 9.03 9.84
11.57 11.94 13.39 14.05
For each model, a plot showing the local scores is visualized. Clicking on the image can enlarge it. If the user only assesses one single model, the absolute quality and absolute local qualities will be provided. We used SVM regression to predict the distance (measured in Angstrom) between the model and the native structure for each residue. We used a 15-residue input window size, and used the following features: amino acid, secondary structure, solvent accessibilities, and contact matrix. For secondary structure and solvent accessibilities, if the predicted one at each residue (predicted from amino acid sequence) is the same as the one parsed (by DSSP) from the model, "1" is used, and otherwise, 0. From the model, we selected all the pairs of residues, with 6 residues away, that have a distance <= 8 Angstrom, and then gathered their predicted probabilities of being contact from our predicted contact map and averaged these probabilities. This averaged probability was inputted as one of the features. To generate the real distance, we use the tool TM_score to align the model with the native. We selected 30 single-domain target proteins in CASP8 to generate training data. These targets contain all the FM single-domain targets and both TBM and HA single-domain targets. The reason we only used single-domain proteins is because we do not want the disorientation of multiple domains to mislead our data. For each of the 30 targets, all of the complete CASP9 TS models were used to generate training examples. Because we were dealing with local qualities, this procedure generated enough examples for us: in total ~180,000 training examples. 5-fold cross-validation was performed and SVM parameters were optimized. The accuracy we got from 5-fold cross-validation is: on the residues that have a real distance with the native <= 10 Angstrom and 20 Angstrom, the average absolute difference between our predicted distance to the actual distance is 1.79 Angstrom and 2.34 Angstrom, respectively. After that, we did a blind test on all the single-domain targets in CASP9, which contained 92 targets. They generated ~800,000 examples. On this CASP9 dataset, if we considered the residues that have a real distance with the native <= 10 Angstrom and 20 Angstrom, the average absolute difference between our predicted value to the real value is 2.60 Angstrom and 3.18 Angstrom, respectively.
APOLLO also outputs a link showing the predicted secondary structures, solvent accessibilities, and contact map used to predict the absolute scores. The first line of the file is the ID, the second line is the amino acid, for example "GSGMKEFPCWLVEEFVVAEECSPCSNFRAKTTPECGPTGYVEKITCSSSKRNEFKSCRSALMEQR". The line below it is the secondary structures, for example "CCCCCCCCCCEEEEEEEEEECCCCCCCEEEECCCCCCCCEEEEEEECCCCCCCCCCCCCHHHHCC". Then it is the solvent accessibilities, for example "eee-ee-e--eeee-e--ee-ee-ee-eeee-ee-eee---ee-e-eeeeeee-ee-eeeeeeee ", and the the contact map matrix, showing the probabilities of being contact for each pair of residues.
FAQs
1. I submit a pool of models, but why the absolute scores for some models are missing?
All the models must contain the exactly same amino acid sequence. If one model has different amino acid sequence, it will not be assigned an absolute quality score. For example, if you submit the tarball here, which contains 8 models. However, one of the model named "FFAS03_TS1" will not be assigned scores. This is because this model contains 59 amino acid, while the other models contain 65 amino acids, i.e., the model "FFAS03_TS1" is a partial model. However, if you really want to evaluate its quality, you can submit it independently as a single model, then APOLLO will assign it an absolute quality score.
2. I only have several models, should I trust the pairwise comparison based scores?
No. If your model number is below 20, we would not recommend you to rely on the scores generated by pairwise comparisons. Pairwise comparison based methods work best when the number of models reaches a large number, for example 300 in CASP9, and when the models are generated by different methods.
3. Are the pariwise comparison scores comparable with the ones I got from another submission?
No. It is only comparable with current submission, i.e., it is related to the other models submitted at the same time. If you change the model pool, the pairwise-based scores are not comparable. But absolute score is.
4. Can I submit a PDB native structure file?
Technically, yes. APOLLO can evaluate protein structures, or models, as long as they are saved in PDB format. So it can evaluate a native structure directly downloaded from PDB. However, the users must make sure that the native structure PDB file only contain one chain. Otherwise, it may cause problem when generating absolute score, and the scores generated by pairwise comaprision are not accurate neither.
5. What is the cut-off absolute value for a good / bad model?
There is no strict cut-off value for a good or bad model. But usually, if a model with a predicted score >= 0.5, it should have a good quality. For the absolute global quality score, if it reaches 0.6, it can be a very good model.
6. Do all the models in the multiple-model tarball must have the same length?
The models one user submitted can have different sequence, as long as they are for the same target protein. Although we did not ask users to input query amino acid sequence, we parse the sequence from each of the models submitted, and select the longest one as the reference sequence. During our pair-wise comparison, we normalize the GDT-TS based on the reference sequence. The reason we suggest users to submit models with the same length is for absolute scores. Because our features were generated on the reference sequence, so if a model's sequence is different to the reference sequence, no absolute score will be generated. One way to solve that is to generate features for each individual model in the model pool. However, that would significantly increase execution time. Also, if the user wants, he/she can always get the absolute score for the interested model by only submitting that single model.
Contact:
If you have any comments or questions, you are welcome to contact Dr. Jianlin Cheng at chengji@missouri.edu or Zheng Wang at zwyw6@mail.missouri.edu.